
Project Computational Materials Science based on papers in
RCR 2024
Digital Discovery 2023
npjCM 2023
JACS 2022
Digital Discovery 2022
Chemical Science 2021
Chemistry of Materials 2020
- Computational Materials Science in our group studies materials and molecules by geometric methods.
- The subarea called Lattice Geometry studies moduli spaces of simpler periodic lattices in low dimensions 2 and 3.
- The related area of Cloud Isometry Spaces studies geometry of moduli spaces of finite clouds of unlabeled points.
- The wider area of Periodic Geometry studies moduli spaces of general periodic point sets that model all periodic crystals.
- The even wider area of Geometric Data Science studies moduli spaces of any data objects up to practical equivalences.
- The latest developments are discussed in the MIF++ seminar and at the annual conference MACSMIN since 2020.
Data-driven analysis of bottle packaging

- DOI : doi:10.1016/j.resconrec.2024.107538
- Abstract. The packaging industry faces mounting demand to integrate post-consumer recyclate (PCR). However, the complex structure-property relationships of PCRs often obscure their performance compared to virgin equivalents, posing challenges in selecting suitable PCRs for applications. Focused on extrusion blow moulding grade high-density polyethylene (HDPE), this study presents the most extensive characterisation of HDPE PCR to date, encompassing 23 resins (3 virgin, 20 PCR). Employing Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), rheology, colour analysis, and mechanical testing, we established a feature-rich dataset with 56 distinctive characteristics. Utilising a data science approach based on principal component analysis, with the virgin samples as a benchmark, we identified that combining FTIR, TGA and mechanical testing provided effective identification of PCRs that closely match the properties of virgin HDPE. The pipeline created can be utilised for new PCRs to determine suitability as a replacement for virgin plastic in a desired application.
@article{smith2024, title={A data-driven analysis of HDPE post-consumer recyclate for sustainable bottle packaging}, author={Philip Smith and Andy McLauchlin and Tom Franklin and Peiyao Yan and Emily Cunliffe and Tom Hasell and Vitaliy A Kurlin and Colin Kerr and Jonathan Attwood and Michael P Shaver and Tom P McDonald}, journal={Resources, Conservation and Recycling}, volume={205}, pages={107538}, year={2024} }
Back to Top of this page | Back to Research & papers | Back to Home page
Invariant-based structural maps of zeolites
 |
|
- DOI : doi:10.1039/D3DD00134B
- Abstract. Zeolites are inorganic materials known for their diversity of applications, synthesis conditions, and resulting polymorphs. Although their synthesis is controlled both by inorganic and organic synthesis conditions, computational studies of zeolite synthesis have focused mostly on the design of organic structure-directing agents (OSDAs). In this work, we combine distances between crystal structures and machine learning (ML) to create inorganic synthesis maps in zeolites. Starting with 253 known zeolites, we show how the continuous distances between frameworks reproduce inorganic synthesis conditions from the literature without using labels such as building units. An unsupervised learning analysis shows that neighboring zeolites according to two different representations often share similar inorganic synthesis conditions, even in OSDA-based routes. In combination with ML classifiers, we find synthesis-structure relationships for 14 common inorganic conditions in zeolites, namely Al, B, Be, Ca, Co, F, Ga, Ge, K, Mg, Na, P, Si, and Zn. By explaining the model predictions, we demonstrate how (dis)similarities towards known structures can be used as features for the synthesis space, thus quantifying the intuition that similar structures often share inorganic synthesis routes. Finally, we show how these methods can be used to predict inorganic synthesis conditions for unrealized frameworks in hypothetical databases and interpret the outcomes by extracting local structural patterns from zeolites. In combination with OSDA design, this work can accelerate the exploration of the space of synthesis conditions for zeolites.
@article{schwalbe2023inorganic, title={Inorganic synthesis-structure maps in zeolites with machine learning and crystallographic distances}, author={Daniel Schwalbe-Koda and Danieal Widdowson and Tuan Anh Pham and Vitaliy Kurlin}, journal={Digital Discovery}, volume={2}, issue={6}, pages={1911-1924}, year={2023} }
Back to Top of this page | Back to Research & papers | Back to Home page
Experimental lithium solid electrolyte conductivities
 |
|
- DOI : 10.1038/s41524-022-00951-z
- Abstract. The application of machine learning models to predict material properties is determined by the availability of high-quality data. We present an expert-curated dataset of lithium ion conductors and associated lithium ion conductivities measured by a.c. impedance spectroscopy. This dataset has 820 entries collected from 214 sources; entries contain a chemical composition, an expert-assigned structural label, and ionic conductivity at a specific temperature (from 5 to 873 °C). There are 403 unique chemical compositions with an associated ionic conductivity near room temperature (15–35 °C). The materials contained in this dataset are placed in the context of compounds reported in the Inorganic Crystal Structure Database with unsupervised machine learning and the Element Movers Distance. This dataset is used to train a CrabNet-based classifier to estimate whether a chemical composition has high or low ionic conductivity. This classifier is a practical tool to aid experimentalists in prioritizing candidates for further investigation as lithium ion conductors
@article{hargreaves2023database, title={A database of experimentally measured lithium solid electrolyte conductivities evaluated with machine learning}, author={Hargreaves, Cameron J and Gaultois, Michael W and Daniels, Luke M and Watts, Emma J and Kurlin, Vitaliy A and Moran, Michael and Dang, Yun and Morris, Rhun and Morscher, Alexandra and Thompson, Kate and others}, journal={npj Computational Materials}, volume={9}, number={1}, pages={9}, year={2023} }
Back to Top of this page | Back to Research & papers | Back to Home page
Invariant-based materials discovery
 |
|
- DOI : 10.1021/jacs.2c02653
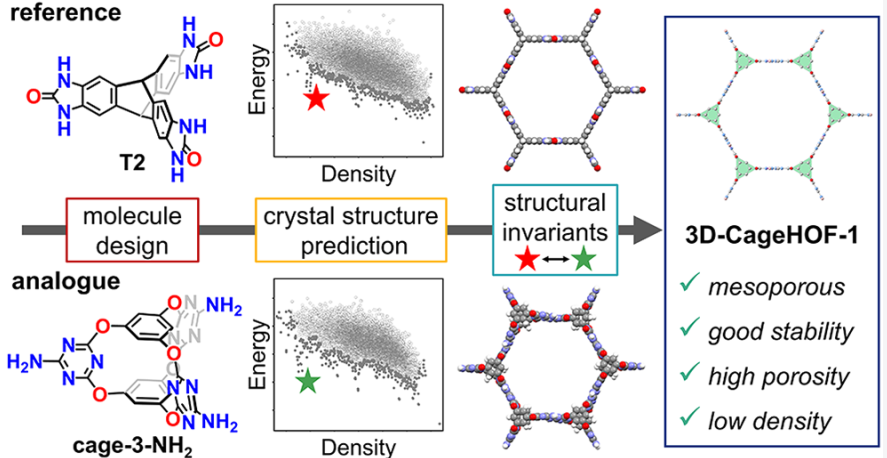
- Abstract. Mesoporous molecular crystals have potential applications in separation and catalysis, but they are rare and hard to design because many weak interactions compete during crystallization, and most molecules have an energetic preference for close packing. Here, we combine crystal structure prediction (CSP) with structural invariants to continuously qualify the similarity between predicted crystal structures for related molecules. This allows isomorphous substitution strategies, which can be unreliable for molecular crystals, to be augmented by a priori prediction, thus leveraging the power of both approaches. We used this combined approach to discover a rare example of a low-density (0.54 g/cm^3) mesoporous hydrogen-bonded framework (HOF), 3D-CageHOF-1. This structure comprises an organic cage (Cage-3-NH2) that was predicted to form kinetically trapped, low-density polymorphs via CSP. Pointwise distance distribution structural invariants revealed five predicted forms of Cage-3-NH2 that are analogous to experimentally realized porous crystals of a chemically different but geometrically similar molecule, T2. More broadly, this approach overcomes the difficulties in comparing predicted molecular crystals with varying lattice parameters, thus allowing for the systematic comparison of energy–structure landscapes for chemically dissimilar molecules.
@article{zhu2022analogy, title={Analogy Powered by Prediction and Structural Invariants: Computationally-Led Discovery of a Mesoporous Hydrogen-Bonded Organic Cage Crystal}, author={Qiang Zhu and Jay Johal, Daniel Widdowson and Zhongfu Pang and Boyu Li and Christopher Kane and Vitaliy Kurlin and Graeme Day and Marc Little and Andrew Cooper}, journal={Journal of the American Chemical Society}, volume={144}, pages={9893–9901}, year={2022} }
Back to Top of this page | Back to Research & papers | Back to Home page
Co-crystals in the Cambridge Structural Database
 |
|
- DOI : 10.1039/D2DD00068G
- Abstract. In this paper we introduce Molecular Set Transformer, a Pytorch-based deep learning architecture designed for solving the molecular pair scoring task whilst tackling the class imbalance problem observed on datasets extracted from databases reporting only successful synthetic attempts. Our models are being trained on all the existing molecular pairs that form cocrystals and are deposited in the Cambridge Structural Database (CSD). Given any new molecular combination, the primary objective of the tool is to be able to select the most effective way to represent the pair and then assign a score coupled with an uncertainty estimation. Molecular Set Transformer is an attention-based framework which learns the important interactions in the various molecular combinations by trying to reconstruct its input by minimizing its bidirectional loss. Several methods to represent the input were tested, both fixed and learnt, with the Graph Neural Network (GNN) and the Extended-Connectivity Fingerprints (ECFP4) molecular representations to perform best showing an overall accuracy higher than 75% on previously unseen data. The trustworthiness of the models is enhanced by adding uncertainty estimates which aims to help chemists prioritize at the early materials design stage both the pairs with high scores and low uncertainty and pairs with low scores and high uncertainty. Our results indicate that the method can achieve comparable or better performance on specific APIs for which the accuracy of other computational chemistry and machine learning tools is reported in the literature. To help visualize and get further insights of all the co-crystals deposited in CSD, we developed an interactive browser-based explorer. An online Graphical User Interface has also been designed for enabling the wider use of our models for rapid in-silico co-crystal screening reporting the scores and uncertainty of any user given molecular pair.
@article{vriza2022molecular, title={Molecular Set Transformer: Attending to the co-crystals in the Cambridge Structural Database}, author={Aikaterini Vriza and Ioana Sovago and Daniel Widdowson and Peter Wood and Vitaliy Kurlin and Matthew Dyer}, journal={Digital Discovery}, volume={1}, issue={6}, pages={834-850}, year={2022} }
Back to Top of this page | Back to Research & papers | Back to Home page
One class classification for co-crystal discovery
 |
|
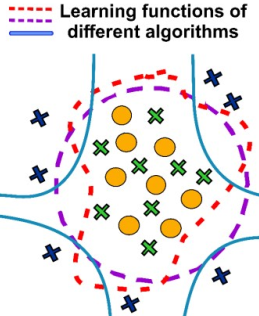
- Abstract. The implementation of machine learning models has brought major changes in the decision-making process for materials design. One matter of concern for the data-driven approaches is the lack of negative data from unsuccessful synthetic attempts, which might generate inherently imbalanced datasets. We propose the application of the one-class classification methodology as an effective tool for tackling these limitations on the materials design problems. This is a concept of learning based only on a well-defined class without counter examples. An extensive study on the different one-class classification algorithms is performed until the most appropriate workflow is identified for guiding the discovery of emerging materials belonging to a relatively small class, that being the weakly bound polyaromatic hydrocarbon co-crystals. The two-step approach presented in this study first trains the model using all the known molecular combinations that form this class of co-crystals extracted from the Cambridge Structural Database (1722 molecular combinations), followed by scoring possible yet unknown pairs from the ZINC15 database (21736 possible molecular combinations). Focusing on the highest-ranking pairs predicted to have higher probability of forming co-crystals, materials discovery can be accelerated by reducing the vast molecular space and directing the synthetic efforts of chemists. Further on, using interpretability techniques a more detailed understanding of the molecular properties causing co-crystallization is sought after. The applicability of the current methodology is demonstrated with the discovery of two novel co-crystals, namely pyrene-6H-benzo[c]chromen-6-one (1) and pyrene-9,10-dicyanoanthracene (2).
@article{vriza2021one, title={One class classification as a practical approach for accelerating pi-pi co-crystal discovery}, author={Vriza, A and Canaj, A and Vismara, R and Cook, L and Manning, T and Gaultois, M and Wood, P and Kurlin, V and Berry, N and Dyer, M and Rosseinsky, M}, journal={Chemical Science}, volume={12}, number={5}, pages={1702--1719}, year={2021} }
Back to Top of this page | Back to Research & papers | Back to Home page
The Earth Mover’s Distance for Inorganic Compositions
 |
|
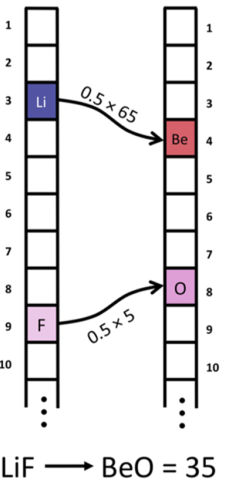
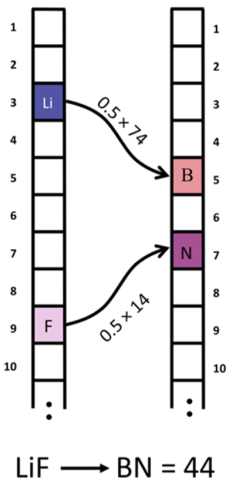
- Abstract. It is a core problem in any field to reliably tell how close two objects are to being the same, and once this relation has been established, we can use this information to precisely quantify potential relationships, both analytically and with machine learning (ML). For inorganic solids, the chemical composition is a fundamental descriptor, which can be represented by assigning the ratio of each element in the material to a vector. These vectors are a convenient mathematical data structure for measuring similarity, but unfortunately, the standard metric (the Euclidean distance) gives little to no variance in the resultant distances between chemically dissimilar compositions. We present the earth mover’s distance (EMD) for inorganic compositions, a welldefined metric which enables the measure of chemical similarity in an explainable fashion. We compute the EMD between two compositions from the ratio of each of the elements and the absolute distance between the elements on the modified Pettifor scale. This simple metric shows clear strength at distinguishing compounds and is efficient to compute in practice. The resultant distances have greater alignment with chemical understanding than the Euclidean distance, which is demonstrated on the binary compositions of the inorganic crystal structure database. The EMD is a reliable numeric measure of chemical similarity that can be incorporated into automated workflows for a range of Machine Learning (ML) techniques. We have found that with no supervision, the use of this metric gives a distinct partitioning of binary compounds into clear trends and families of chemical property, with future applications for nearest neighbor search queries in chemical database retrieval systems and supervised ML.
@article{hargreaves2020earth, title={The Earth Mover’s Distance as a Metric for the Space of Inorganic Compositions}, author={Hargreaves, Cameron J and Dyer, Matthew S and Gaultois, Michael W and Kurlin, Vitaliy A and Rosseinsky, Matthew J}, journal={Chemistry of Materials}, volume={32}, issue={24}, pages={10610-10620}, year={2020} }
Back to Top of this page | Back to Research & papers | Back to Home page